
 48炒鸡内金配方颗粒.docx
48炒鸡内金配方颗粒.docx
《48炒鸡内金配方颗粒.docx》由会员分享,可在线阅读,更多相关《48炒鸡内金配方颗粒.docx(4页珍藏版)》请在第一文库网上搜索。
1、辽宁省药品监督管理局中药配方颗粒标准1NPFK1-2023048炒鸡内金配方颗粒ChaojineijinPeifangke1i【来源】本品为雉科动物家鸡Ga11usga11usdomesticusBrisson的干燥沙囊内壁经炮制并按标准汤剂的主要质量指标加工制成的配方颗粒。【制法】取炒鸡内金饮片8000g,加水煎煮,滤过,滤液浓缩成清膏(干浸膏出膏率为4%8%),加入辅料适量,干燥(或干燥,粉碎),再加入辅料适量,混匀,制粒,制成IoOOg,即得。【性状】本品为黄白色至棕黄色的颗粒;气微腥,味苦。【鉴别】(I)取本品1.5g,研细,加乙醇30m1,加热回流30分钟,滤过,滤液蒸干,残渣加水2
2、0m1使溶解,用三氯甲烷振摇提取2次,每次15m1,合并三氯甲烷液,蒸干,残渣加甲醇Im1使溶解,作为供试品溶液。另取鸡内金对照药材0.2g,加水IOm1煮沸30分钟,滤过,滤液蒸干,残渣自“加乙醇30m1”起,同法制成对照药材溶液。照薄层色谱法(中国药典2023年版通则0502)试验,吸取供试品溶液81对照药材溶液151,分别点于同一硅胶G薄层板上,以三氯甲烷-乙酸乙酯-甲酸(3:3:0.5)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105C加热至斑点显色清晰,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。(2)取本品0.1g,
3、研细,力口1%碳酸氢镂溶液25m1,超声处理30分钟,用微孔滤膜滤过,取续滤液1m1,加胰蛋白酶溶液1001(取序列分析用胰蛋白酶,加1%碳酸氢镀溶液制成每ImI中含Img的溶液,临用时配制),摇匀,37恒温酶解12小时,作为供试品溶液。另取鸡源多肽I对照品、鸡源多肽H对照品适量,精密称定,力口1%碳酸氢核溶液制成每Im1各含2g的混合溶液,作为对照品溶液。照高效液相色谱法-质谱法(中国药典2023年版通则0512和通则0431)试验,以十八烷基硅烷键合硅胶为填充剂(柱长为IOOmm,内径为2.1mm,粒径为1.7m1.9m);以乙懵为流动相A,以0.1%甲酸溶液为流动相B,按下表中的规定进行
4、梯度洗脱,流速为每分钟0.3m1。采用质谱检测器,电喷雾正离子模式(ESI+),进行多反应监测(MRM),选择质荷比(z)379.21(双电荷)一571.36和m/z379.21(双电荷)385.26,m/z785.41(双电荷)941.51和m/z785.41(双电荷)245.08作为检测离子对进行检测。取对照品溶液,进样21,按上述检测离子对测定的MRM色谱峰的信噪比均应大于3:1。时间(分钟)流动相A(%)流动相B(%)0582092805-102035806510-113590651011-139010吸取供试品溶液21,注入高效液相色谱.质谱联用仪,测定。以质荷比(m/z)379.2
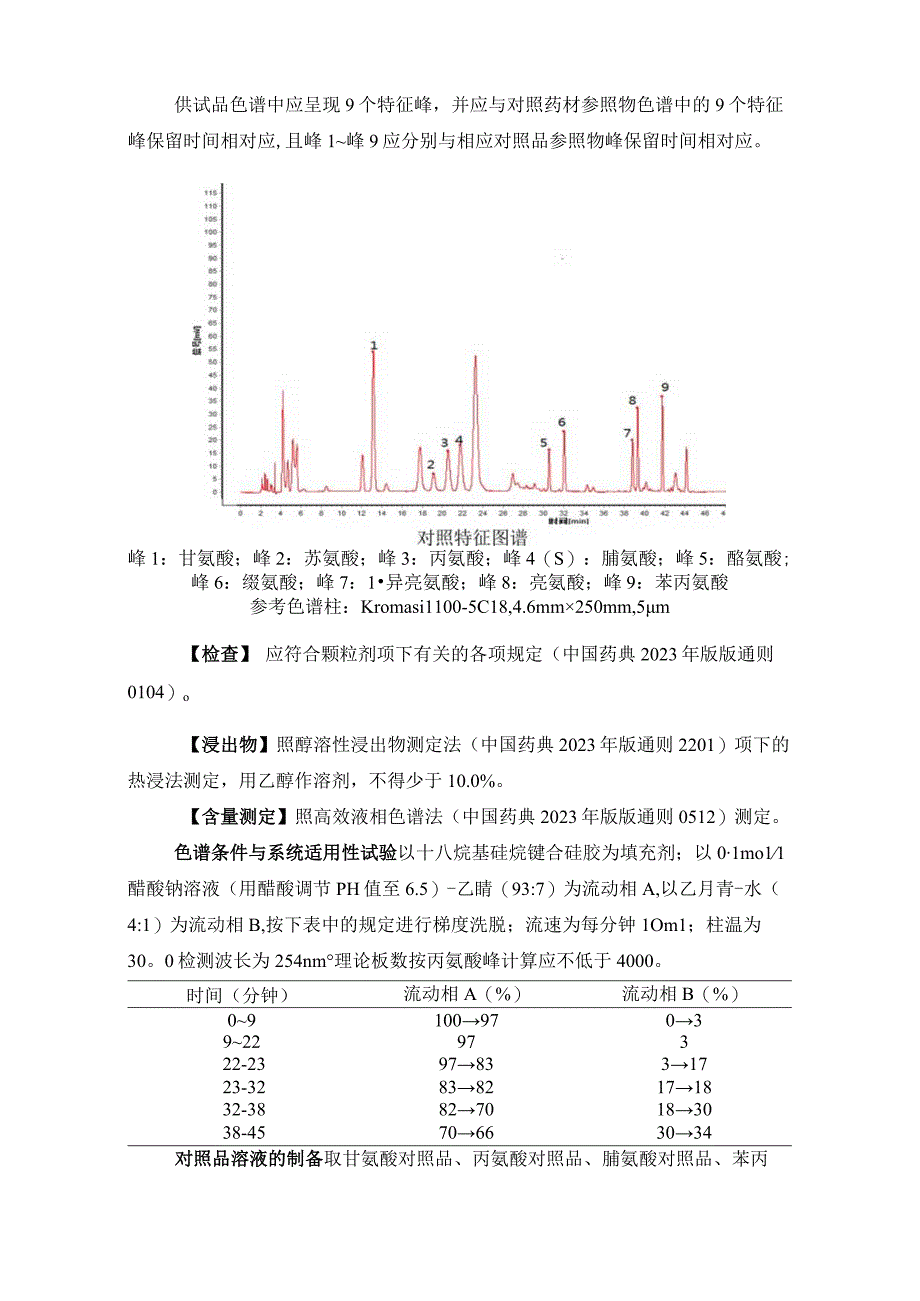
5、1(双电荷)-571.36和m/z379.21(双电荷)-385.26,m/z785.41(双电荷)941.51和mz785.41(双电荷)-245.08离子对提取的供试品离子流色谱中,应同时呈现与对照品色谱保留时间一致的色谱峰。【特征图谱】照高效液相色谱法(中国药典2023年版版通则0512)测定。色谱条件与系统适用性试验同【含量测定】项。参照物溶液的制备取鸡内金对照药材0.1g,置具塞水解管中,加入6mo11盐酸溶液IOm1,密塞,置150C中水解3小时,放冷,滤过,滤液移至蒸发皿中,水解管与滤渣再用水IOm1分次洗涤,滤过,滤液并入蒸发皿中,蒸干,残渣加O.ImoV1盐酸溶液溶解,转移至




- 配套讲稿:
如PPT文件的首页显示word图标,表示该PPT已包含配套word讲稿。双击word图标可打开word文档。
- 特殊限制:
部分文档作品中含有的国旗、国徽等图片,仅作为作品整体效果示例展示,禁止商用。设计者仅对作品中独创性部分享有著作权。
- 关 键 词:
- 48 鸡内金 配方 颗粒
 第一文库网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
第一文库网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 调和油系列产品项目可行性研究报告.doc
调和油系列产品项目可行性研究报告.doc
-
三相调压器项目可行性研究报告.doc
-
可行性分析报告设立XX基金管理公司项目可行性分析报告.doc
-
丁硅料项目可行性研究报告.doc
-
二手车交易市场建设项目可行性研究报告.doc
-
二手车交易市场项目可行性研究报告.doc
-
二万头商品猪项目可行性研究报告大纲2.doc
-
二氢松油醇项目可行性研究报告.doc
-
二氧化钛项目可行性研究报告.doc
-
二位五通电磁换向阀项目可行性研究报告.doc
-
二聚酸项目可行性研究报告.doc
-
从化项目可行性报告XXXX329.doc
-
二氧化碳回收项目可行性研究报告.doc
-
水性油墨工程技术研发中心项目可行性研究报告.doc
-
水杀菌消毒项目可行性研究报告.doc
-
水稻种植及加工产业化项目可行性报告.docx
-
水暖五金配件项目可行性研究报告.doc
-
汽车行业-汽车教学整车模型项目可行性研究报告.doc
-
汽车行业-汽车电机零部件项目可行性研究报告.doc
-
汽车行业-汽车空调压缩机支架项目可行性研究报告.doc
-
汽车行业-汽车轮钢项目可行性研究报告摩森咨询·专业编写可行性.doc
-
水果筛选项目可行性研究报告.doc
-
水质改良剂项目可行性研究报告.doc
-
汽车行业-汽车空调电器总成项目可行性研究报告.doc
-
汽车行业-汽车钢卷项目可行性研究报告.doc
-
水泥厂项目可行性研究报告.docx
-
江苏省某幼儿园学校配套停车场项目建议书(代可行性研究报告).docx
-
水泥预制件墙板项目可行性研究报告.doc
-
江西省金韵生态农业示范园建设项目可行性研究报告页(1).docx
-
汽车行业-汽车美容护理用品项目可行性研究报告.doc
-
汽车行业-汽车轮胎套筒项目可行性研究报告.doc
-
汽车行业-汽车阀门项目可行性研究报告.doc
-
3篇2023主题教育以学铸魂以学增智以学正风以学促干研讨发言感悟心得体会.docx
-
3篇人事变动会议记录干部提拔调整.docx
-
3涵洞工程工程文档范本.docx
-
3篇街道社会心理服务体系建设工作状况汇报.docx
-
3马致远《双调寿阳曲·人初静》题解公开课教案教学设计课件资料.docx
-
广西瑞丰化工有限公司年产1万吨环保塑料包装制品及1万只高档彩色纸箱生产基地项目环评报告.doc
-
长乐页岩砖厂仿古砖生产线技术改造项目环评报告.doc
-
3篇2023年六个必须坚持党员干部学习心得体会感想附党课讲稿.docx
-
3马致远《〔南吕〕四块玉·恬退》原文与赏析公开课教案教学设计课件资料1.docx
-
4月职位空缺意外增加.docx
-
桂平市华盈服饰有限公司年产2吨加工染色羽毛项目环评报告.docx
-
广西华佰建材有限公司百色市年产40万吨工业脱硫石膏粉及配套项目环评报告.doc
-
广西远涛投资发展有限公司年产70万平方米钢化玻璃项目环评报告.docx
-
广西贵港茂盛科技有限公司高端智能家俱产业园(一期)项目环评报告.docx
-
清平高速公路二期工程环评报告.doc
-
贵港市城市固废处理循环利用中心项目(变更)环评报告.doc
-
贵港市兴泰木业有限公司(江南园二分厂)年产15万套板式家具建设项目环评报告.doc
-
贵港市溢丰达服装有限公司年产550万件针织服装项目环评报告.doc
-
413 四维联动 巧搭支架公开课教案教学设计课件资料.docx

